
If you are developing a medical device for the U.S. market, you first need to determine how your device will be classified by FDA.
If you do not know the FDA classification, I recommend you go check out the step-by-step guide to regulatory product classification.

MedCity News Pivot Podcast: How Emergency Departments Are Using Real-Time Data to Improve Outcomes
How can AI benefit and improve patient outcome in the emergency department? Watch the latest episode to find out.
In the U.S., a large percentage of medical devices are class II and will require a 510(k) clearance from FDA before going to market.
What is a 510(k) submission?
According to FDA:
A 510(K) is a premarket submission made to FDA to demonstrate that the device to be marketed is at least as safe and effective, that is, substantially equivalent, to a legally marketed device (21 CFR §807.92(a)(3)) that is not subject to premarket approval.


Enhanced Direct Enrollment: Georgia Access and the Power of Partnership
Across the nation, state-based health insurance exchanges are searching for ways to better serve their communities while making enrollment easier, more personal, and more accessible for everyone.
“Substantial equivalence” is a key term. This means you need to compare your product to another medical device, or predicate device, which has already received 510(k) clearance.
Note, if you are preparing a 510(k) submission, you need to check out the FDA’s “Refuse to Accept Policy for 510(k)s”. There is a RTA checklist in this FDA document. I want to provide you with a free, usable version of this checklist.
[Click Here To Access a Free RTA Checklist]
Submitting a 510(k) Without Design Controls – Is This Possible?
Is it possible to draft a 510(k) submission without design controls?
For the past 17 years, I have believed the answer to that question to be an emphatic “no”.
However, in the past few months, I have come across many, many examples of companies and individual who have submitted 510(k)s and documented no design controls.
It blows my mind that somebody would consider submitting a 510(k) without having design controls documented in a design history file.
Why?
As stated by FDA, a 510(k) is about demonstrating your medical device is safe and effective.
Design controls are documented, objective evidence you establish throughout the product development process to prove your device is safe and effective.
To me, design controls actually “feed” into your 510(k) submission.
To me, there is no way to construct a 510(k) submission without design controls.
Did you know that there are actually consultants who do nothing but write 510(k) submissions?
Please beware! Many of these 510(k) consultants do NOT ensure that you have established and documented design controls and a design history file; they just author the 510(k).
Whenever I am tasked with authoring a 510(k) submission, I start first with establishing the design controls and DHF. I actually use the contents of a DHF to guide me in preparing a 510(k) submission.
Documenting and organizing design controls in a DHF should be the first step taken in preparing a 510(k) submission.
Realize this.
Once you get FDA 510(k) market clearance, you will be subject to inspection by FDA. It may not happen immediately after getting clearance. However, you should anticipate that an inspection will happen since FDA has a mandate to do so at least every two years for medical device companies with class II and class III products.
And year after year, design controls are cited as the #1 issue during FDA inspections.
Doesn’t it make logical sense to ensure and establish that design controls are captured and documented before you actually submit that 510(k)?
Do you have a medical device which has received 510(k) market clearance yet lacks design controls? If so, you can and should correct this. Contact me if you want some help figuring out how and what to do.
You should also continue reading to learn what you can do on your own.
How Design Controls Apply to a 510(k)
Let me remind you a bit about design controls.
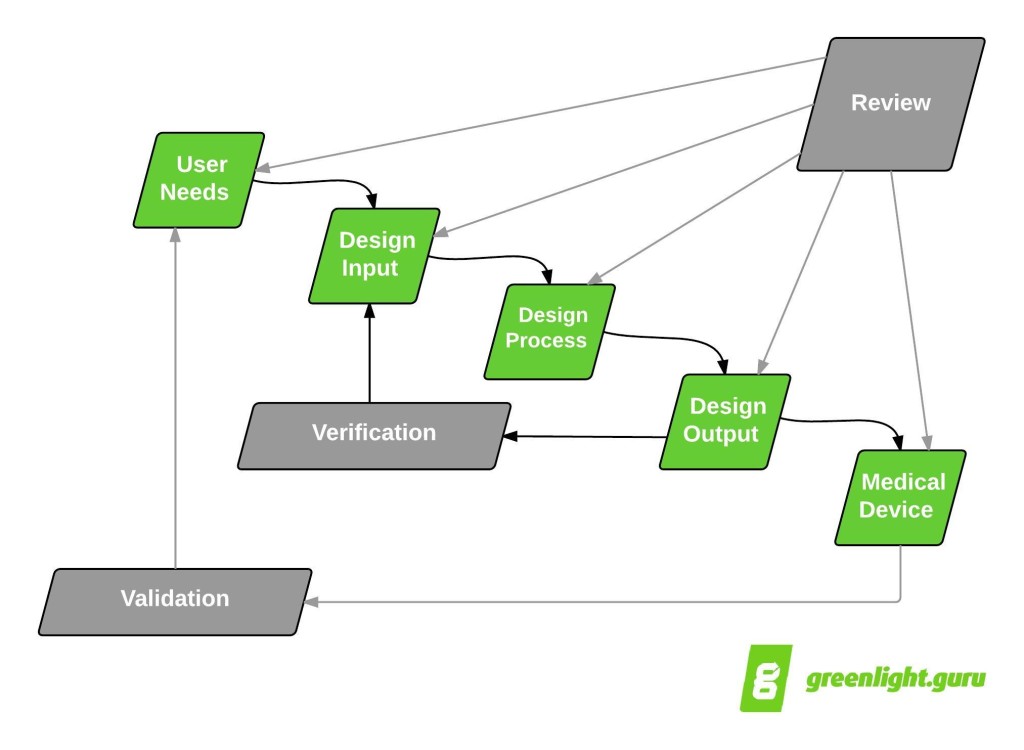
Generally speaking, the time at which you are ready to submit a 510(k) will happen during design verification / design validation stages of product development.
I am going to outline all of the sections of a 510(k) submission and point out when and where your design controls fit into the submission.
First let me start with the contents of a 510(k):
|
Section |
|
1.0 Medical Device User Fee Cover Sheet (Form FDA 3601) |
|
2.0 CDRH Premarket Review Submission Cover Sheet |
|
3.0 510(k) Cover Letter |
|
4.0 Indications for Use Statement |
|
5.0 510(k) Summary |
|
6.0 Truthful and Accurate Statement |
|
7.0 Class III Summary and Certification |
|
8.0 Financial Certification or Disclosure Statement |
|
9.0 Declarations of Conformity and Summary Reports |
|
10.0 Executive Summary |
|
11.0 Device Description |
|
12.0 Substantial Equivalence Discussion |
|
13.0 Proposed Labeling |
|
14.0 Sterilization and Shelf Life |
|
15.0 Biocompatibility |
|
16.0 Software |
|
17.0 Electromagnetic Compatibility and Electrical Safety |
|
18.0 Performance Testing – Bench |
|
19.0 Performance Testing – Animal |
|
20.0 Performance Testing – Clinical |
[Click here to access a free 510(k) table of contents template]
Let me also suggest you check out the “Guidance for Industry and FDA Staff: Format for Traditional and Abbreviated 510(k)s”.
At least 14 of the required 20 sections of a 510(k) relate to your design controls. Below, I will explain the specific sections of a 510(k) submission that apply and relate to design controls.
Section 4.0 Indications for Use Statement
The FDA describes the indications for use statement as:
The statement should include specific indications, clinical settings, define the target population, anatomical sites, etc. This statement must be consistent with your labeling, advertising and instructions for use.
Your indications for use describe the entire purpose for your medical device. This statement then feeds into establishing user needs for your product.
And user needs get the design controls process going. User needs are the primary basis for establishing your design inputs. Towards the end of your development efforts, user needs also play a significant part. Design validation demonstrates the product you have developed meets the user needs.
Section 5.0 510(k) Summary
The 510(k) summary is an overview of your medical device. The 510(k) summary includes:
-
A description of the device according to the product labeling (relates to design outputs)
-
Explanation of how the device functions (relates to design inputs)
-
Physical and performance characteristics including device design, materials, and physical properties (relates to design inputs)
-
Predicate device comparison via technological characteristics and testing (relates to design inputs, design outputs, and design verification)
-
Summary of nonclinical testing (or clinical testing, if applicable)(relates to design verification)
These items are a combination of design inputs, design outputs, and design verification.
Section 9.0 Declarations of Conformity and Summary Reports
For some device types, there may be applicable performance standards to follow during the product development process. Examples include IEC 60601-1 and ISO 10993 (among many, many others).
In this section, you are to provide declarations of conformity to any standards you have followed.
Criteria contained within standards often relates to design inputs as well as design verification.
Section 10.0 Executive Summary
The executive summary section is a culmination of indications for use (section 4.0), device description (section 11.0), device comparison (section 12.0), and performance testing (sections 18.0, 19.0, and 20.0).
There are elements of user needs, design inputs, design outputs, design verification, and design validation included in the executive summary.
Section 11.0 Device Description
The section for device description includes details relating to design inputs and design outputs. This section can often be fairly detailed and include drawings and schematics to help explain your product to FDA.
Section 12.0 Substantial Equivalence Discussion
In this section, you are comparing your medical device to the predicate product.
This substantial equivalence discussion will primarily focus on specific product characteristics (design outputs) and side by side comparison, often in the form of testing (design verification).
Section 13.0 Proposed Labeling
Proposed labeling includes the labels included on your product and packaging, as well as instructions for use and operator manuals.
Labeling should be described and captured as design outputs.
Section 14.0 Sterilization and Shelf Life
Sterilization and shelf life section has applicability to design inputs and design verification.
Design inputs should describe sterilization methods, identify specific sterilization parameters, describe packaging criteria, and shelf life.
And your design verification activities shall demonstrate these inputs have been met. This will include sterilization validation and shelf life testing.
Section 15.0 Biocompatibility
Biocompatibility relates to design inputs and design verification.
Design inputs shall describe type of body contact and duration. Design verification shall demonstrate the materials are safe and effective and meet the established biocompatibility design inputs.
Section 16.0 Software
If your device contains software, this section of your 510(k) shall contain software-related design inputs, design outputs, and design verification activities.
Be prepared to follow the FDA’s “Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices”and provide documentation described within this document.
Section 17.0 Electromagnetic Compatibility and Electrical Safety
If your medical device has electronics, this section shall contain evidence to support electrical safety. This relates to design inputs, design outputs, and design verification.
The big item related to EMC and electrical safety is the IEC 60601 series of standards.
Section 18.0 Performance Testing – Bench
Any “bench” testing conducted as part of product development efforts shall be summarized in this section of your 510(k). This relates to design verification activities.
Section 19.0 Performance Testing – Animal
Any “animal” testing conducted as part of product development efforts shall be summarized in this section of your 510(k). This often relates to design verification activities and sometimes design validation.
Section 20.0 Performance Testing – Clinical
Any “clinical” testing conducted as part of product development efforts shall be summarized in this section of your 510(k). This often relates primarily to design validation.
Jon Speer is the founder and VP of QA/RA at Greenlight Guru, a software company that produces the only modern quality management software solution exclusively for medical device companies. Device makers in hundreds of cities in more than 30 countries use Greenlight Guru to get safer products to market faster while pushing beyond compliance to True Quality.
Jon is a medical device industry veteran with over 20 years experience having helped dozens of devices get to market over his career in a variety of roles including product development, project management, quality and regulatory. He is a thought leader, speaker and regular contributor at numerous leading industry publications. He is also the host of the #1 most downloaded podcast in the industry, The Global Medical Device Podcast.
This post appears through the MedCity Influencers program. Anyone can publish their perspective on business and innovation in healthcare on MedCity News through MedCity Influencers. Click here to find out how.